Actualidad

18/04/2024
Implantar mejoras “con la mayor brevedad posible” orientadas al incremento de recursos y personal; a desplegar medidas de apoyo en salud mental para el conjunto de profesionales; a mejorar la formación continua y el acceso a la innovación, y el reconocimiento y valoración pública de la labor que desarrollan los médicos de familia. Estas son las cuatro demandas que María Ángeles Medina Martínez, presidenta de la Societat Valencia...

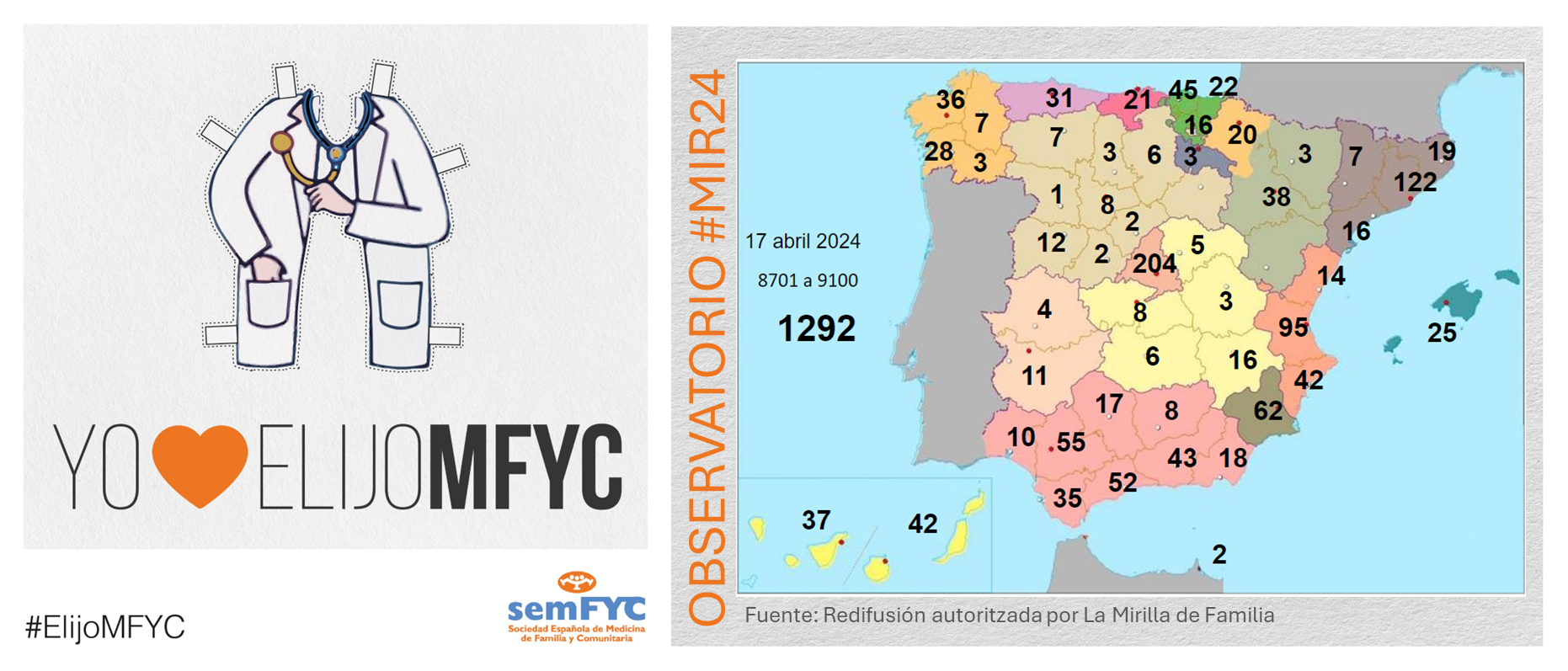
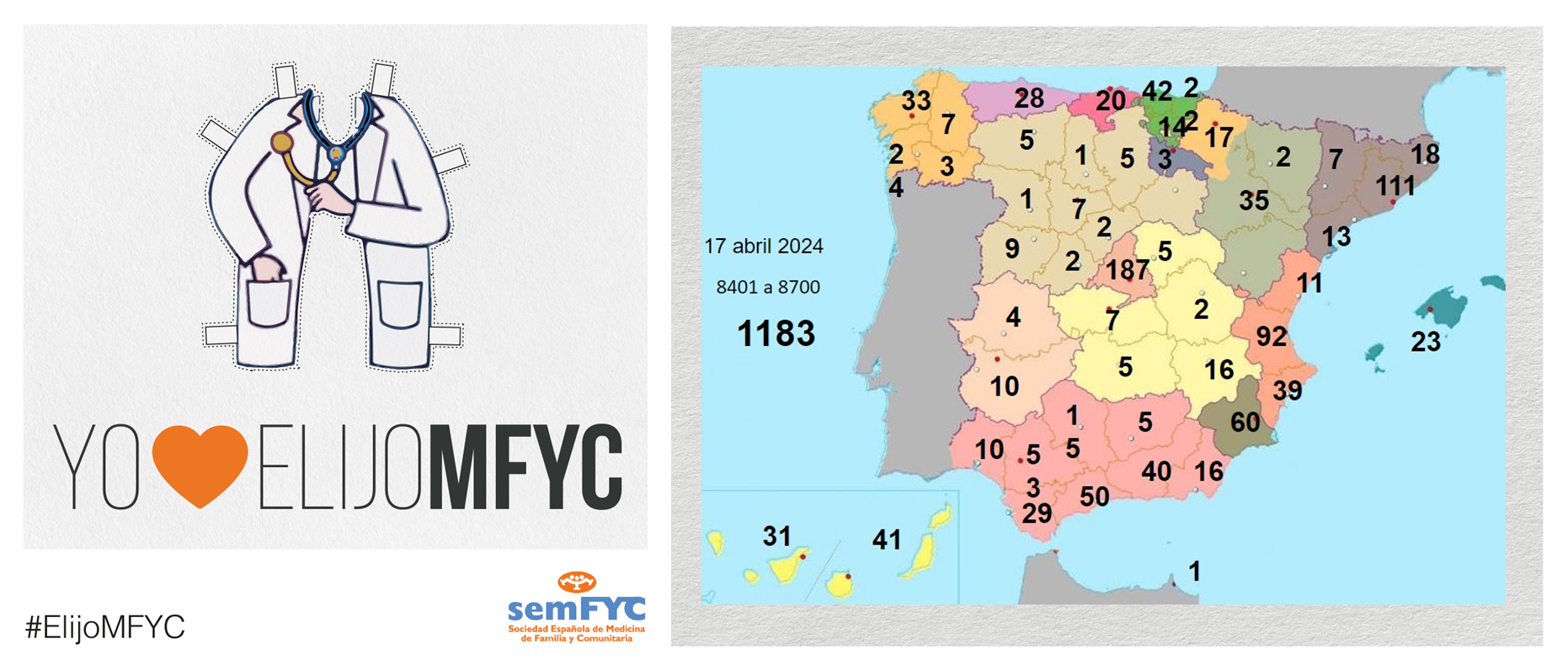
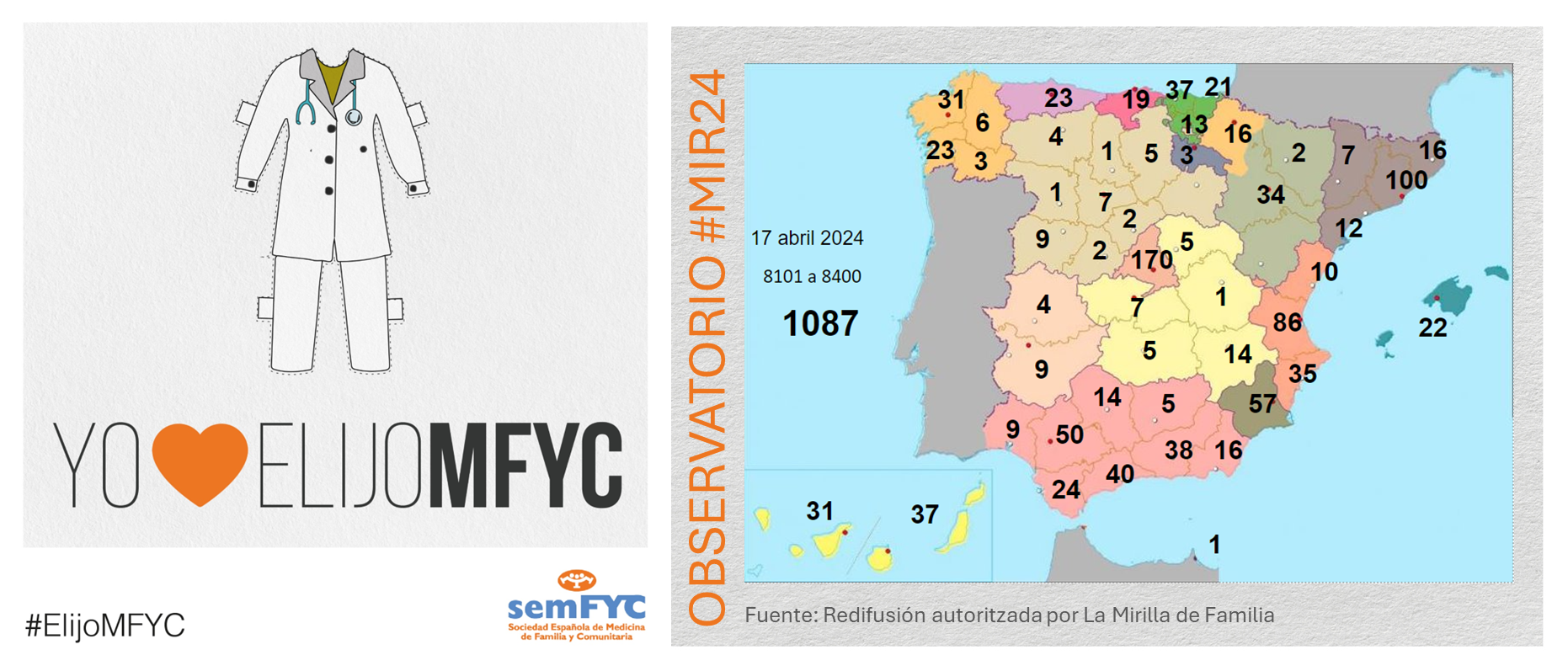
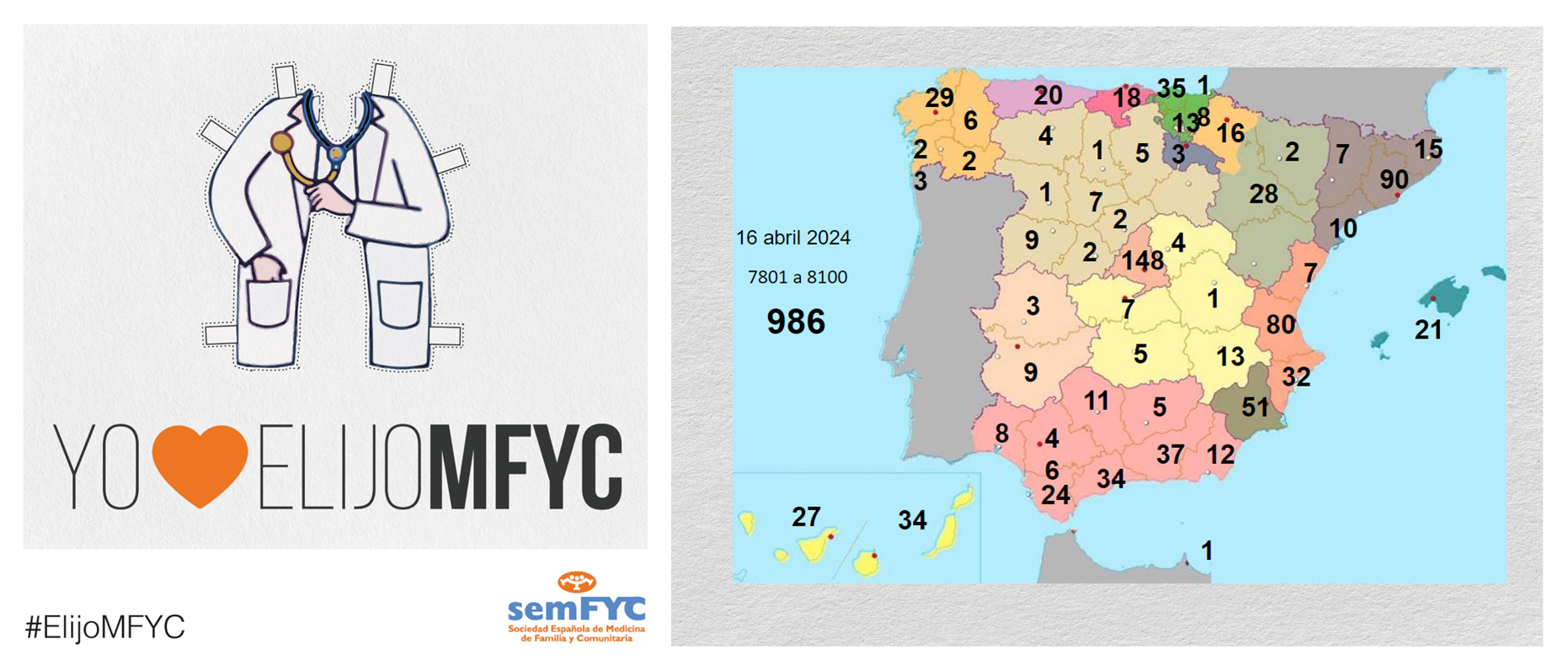

.png )