Actualidad

18/04/2024
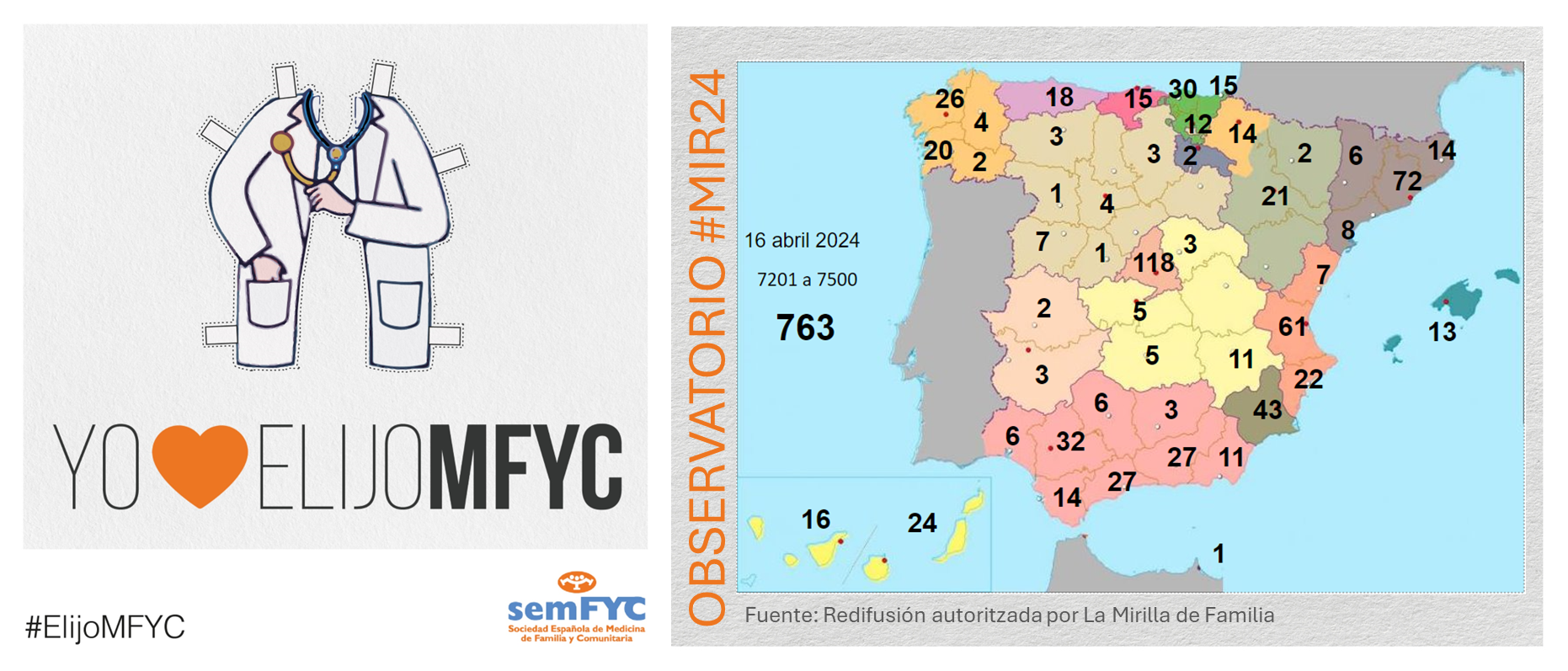
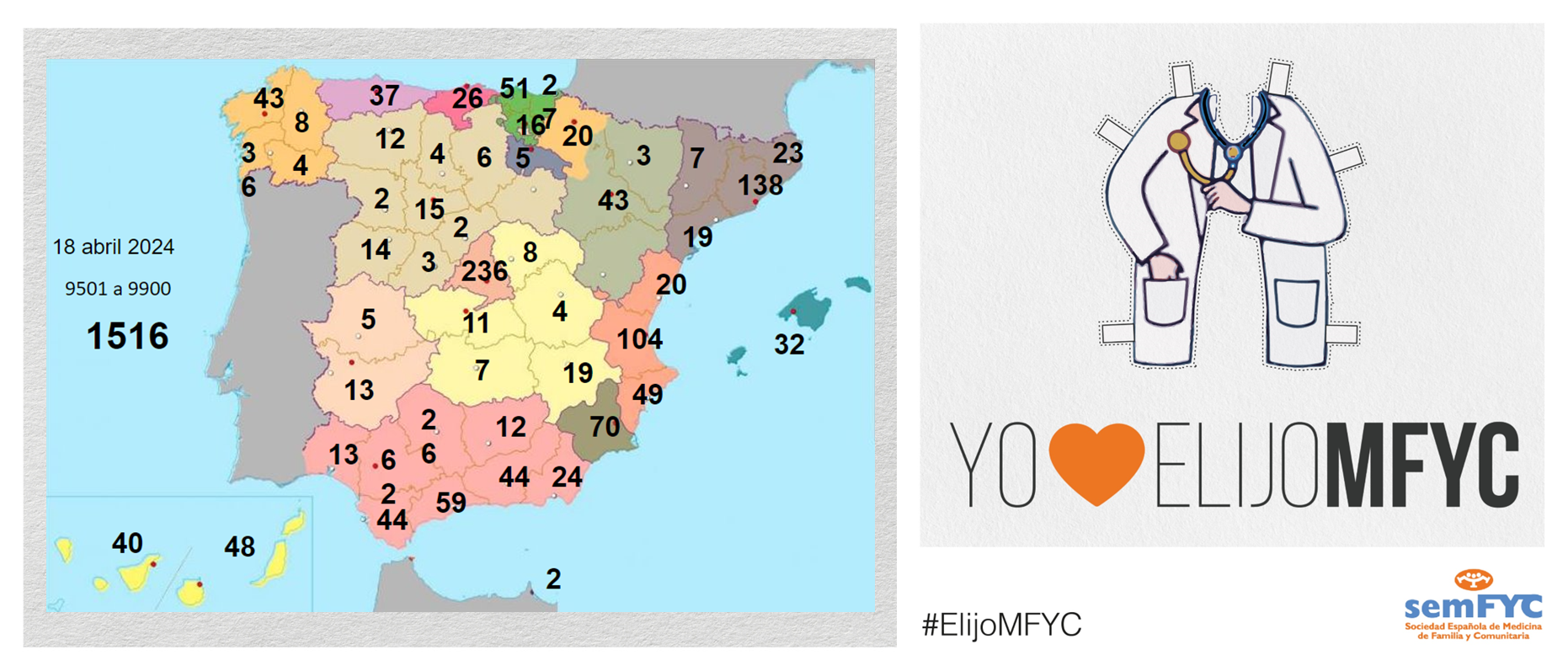
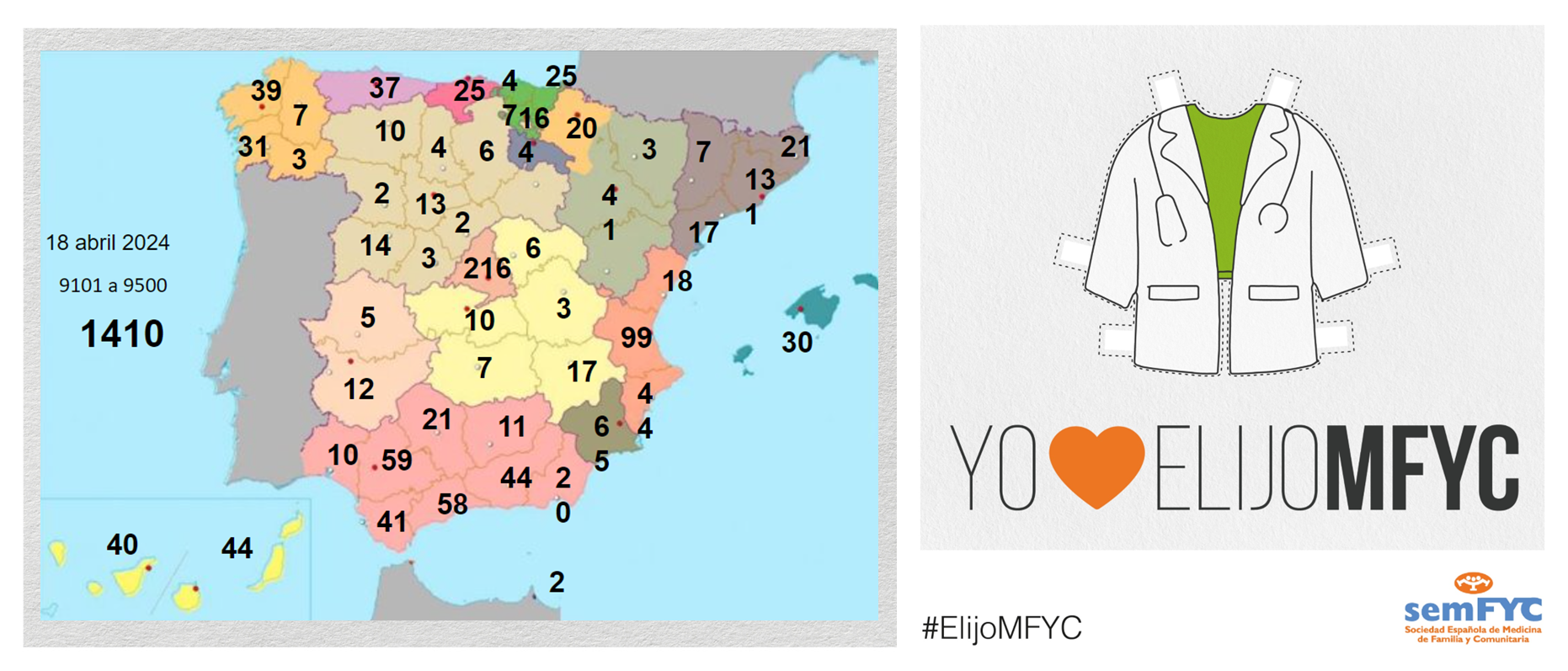
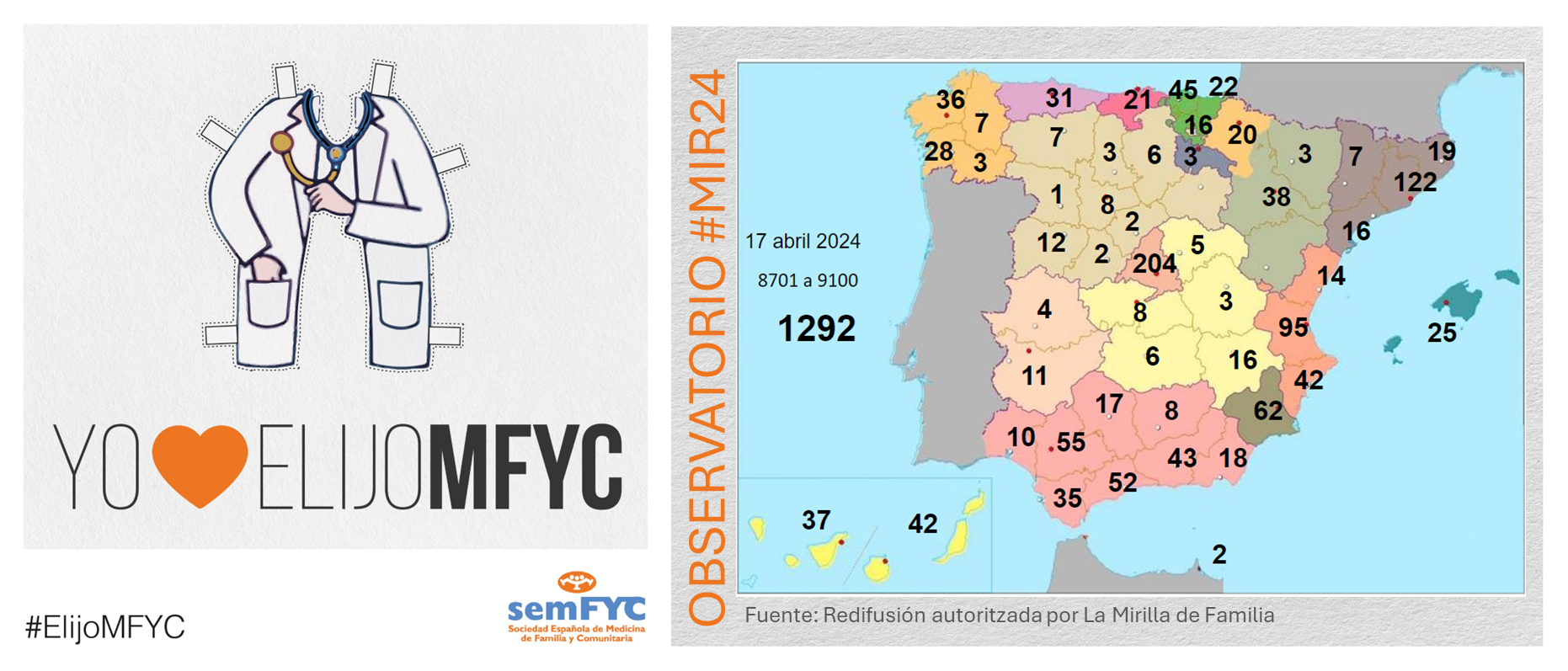
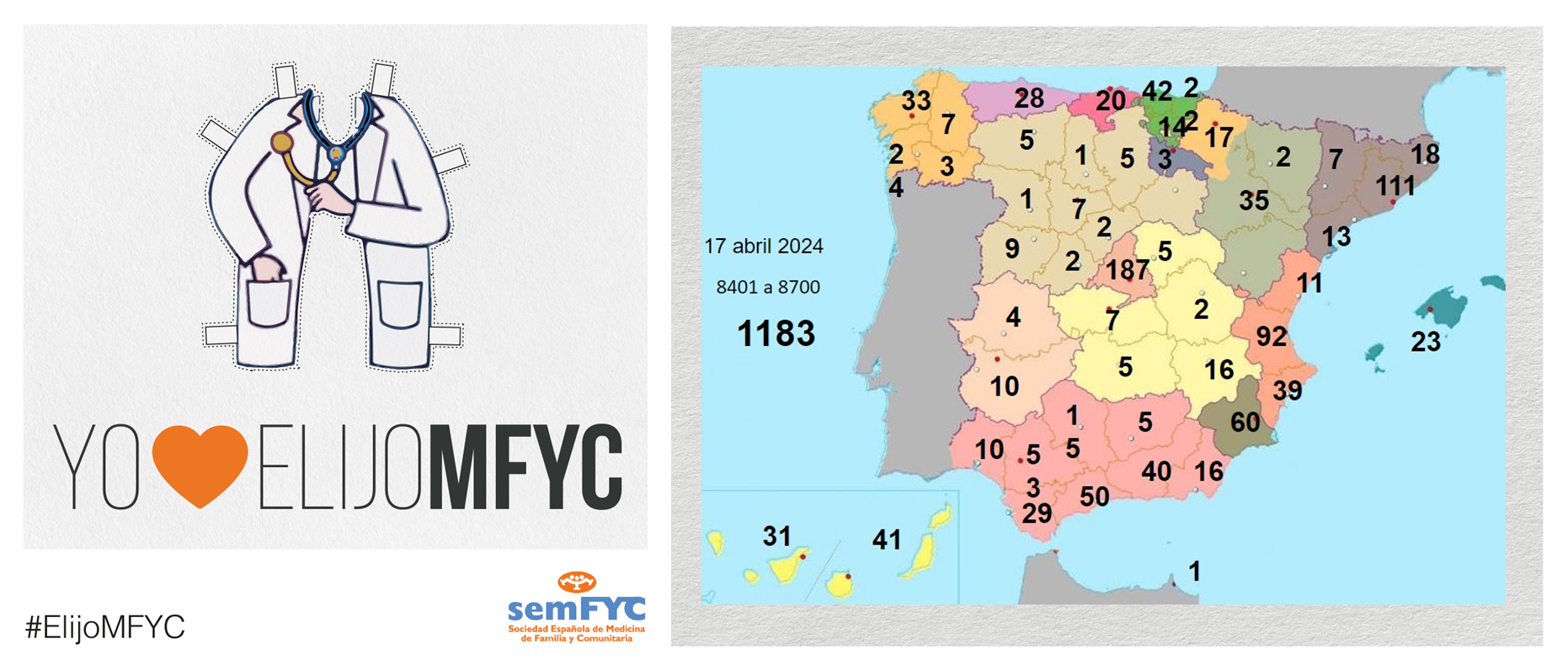
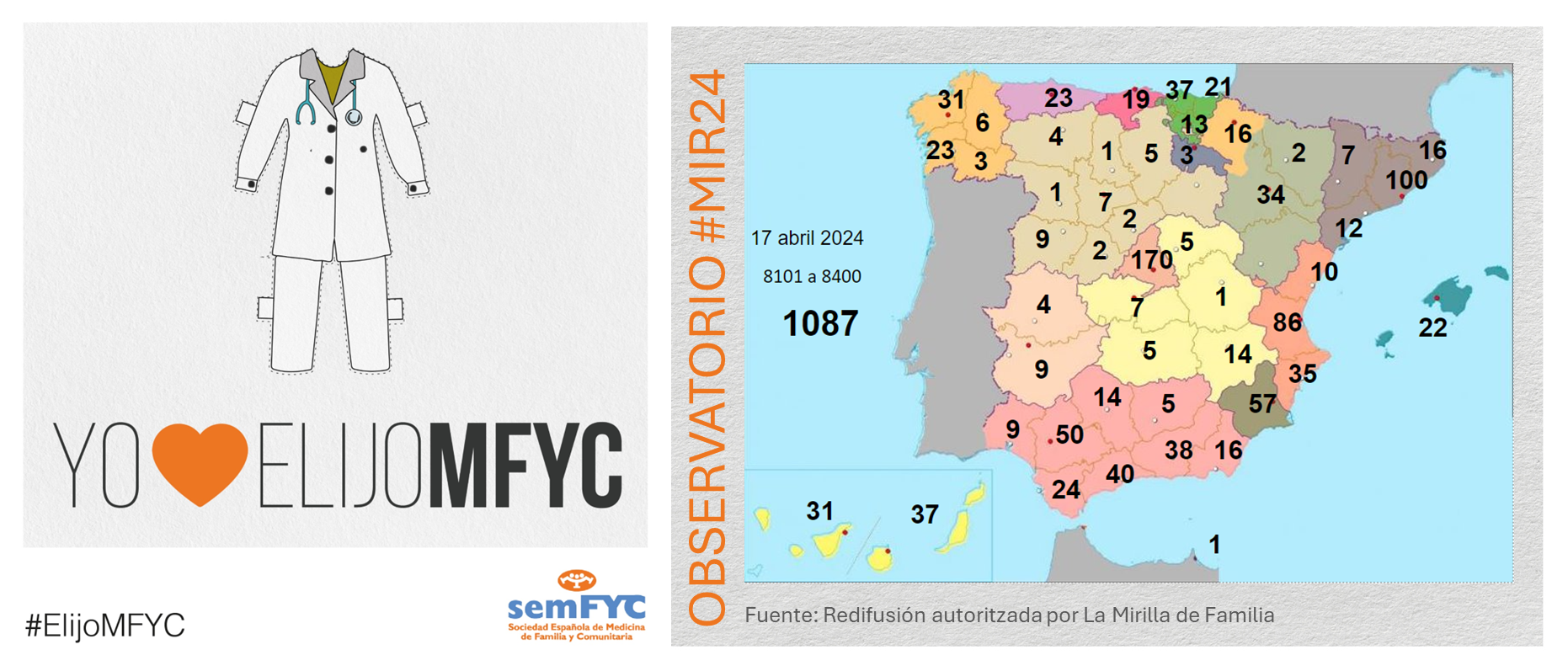
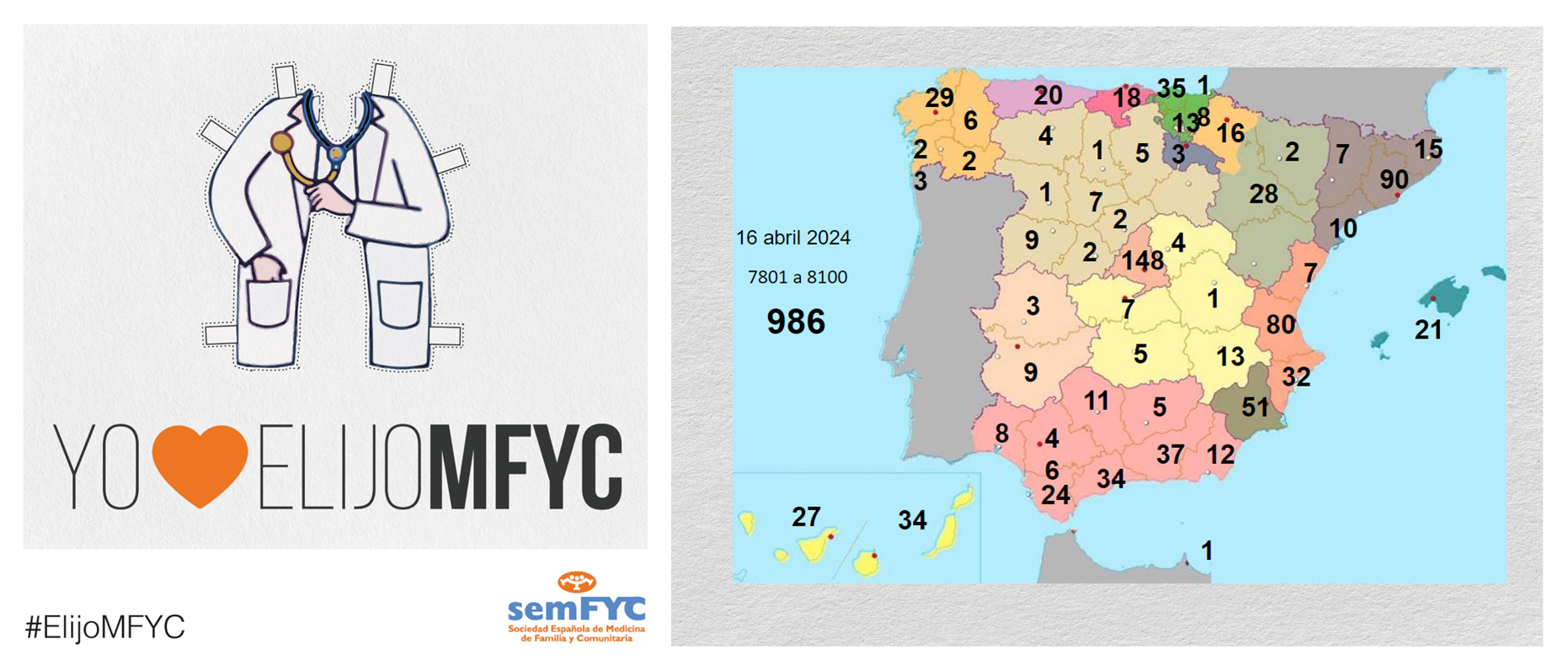
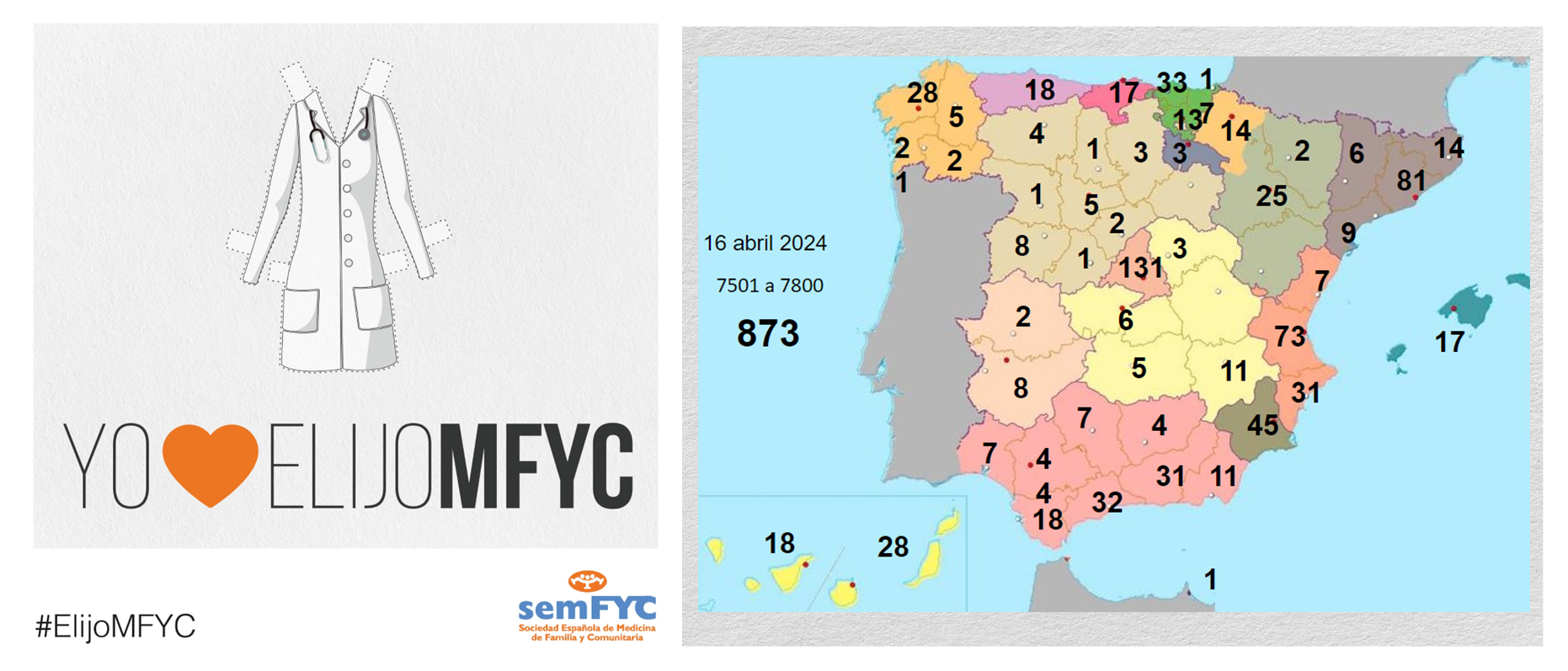
El tercer “round” de la penúltima jornada de elección suma 135 nuevas plazas y con la noticia de que por fin Soria ya cuenta con residentes.
Terminamos el penúltimo día de elección MIR con 135 nuevas plazas asignadas, y con la elección por fin de Soria. Ya solo queda Teruel por tener la posibilidad de relevo.
En cuanto a las comunidades autónomas: Madrid, Canarias y Ceuta completan hoy su oferta de plazas, mientras que en el Pa&iac...



.png )